Poremećaje razvoja noći i periferni vid

sadržaj
stacionarni poremećaj
Kongenitalna stacionarni noćno slijepilo
Kada je patologija promatrana neprogresivni noćno sljepilo (dan-sljepoća), i tamna prilagodbu, koja je uzrokovana šipki-System funkcije poremećaj je genetički heterogena bolest. Možda nasljeđivanja autosomno dominantno ili recesivno tipa, i nasljeđe vezan na X kromosomu. Razvoj bolesti je povezano s mutacijom gena koji kodiraju Postupak sinteze, transport i skladištenje rhodopsina,
Više različitih oblika bolesti.
1. Kongenitalna stacionarni noći sljepoća normalno oko dna:
- Vrsta Riggs (I tipa);
- tip-Schubert Bornstein (tip II) - postoje dva podtipa: potpuna disfunkcija šipke-sustava i njegove rezidualna funkcija.
2. Kongenitalna stacionarni noći sljepoća s promjenama fundusa:
- Ogushi bolest - I i tipa II;
- belotochechnoe eyeground;
- Pjegavi mrežnica iskrenosti.
Očni simptomi. Smanjena amplitudu dva vala. Ritmička ERG na 30 Hz frekvencija ne mijenja. Krivulja tamno prilagodbe monofaznu slobodan od stošca šipkastog zavoja. Vidna oštrina obično nije promijenilo, rijetko blago smanjena. Karakteristike rodopsinu prema reflektometrije su normalni. Bolest se nasljeđuje u recesivnim sex-linked tipa.
Kongenitalna stacionarna noći sljepoća normalno oko tipa II dno (Schubert-Bornstein) karakterizira nedostatak, odnosno znatno smanjenje b-valova ERG oba tipa pod normalnim ili povećane amplitude od vala ( „negativna negativne” ERG).
Uz punu tipu kongenitalne stacionarni noći sljepoća Schubert-Bornstein tamni ERG odsutnog Photopic - spremljenu ili subnormalnom. Maksimalna ERG umanjena negativan. Tamno adaptacija dramatično umanjena. U kombinaciji sa srednjim i visokim myopia mjeri ambliopije i nistagmus.
Nepotpuna vrsta kongenitalne stacionarni noći sljepoća Schubert-Bornstein je povećanje u tamnom adaptaciji pragovi 1,0-1,5 zapisnik. U Tamni ERG smanjena za maksimalno ERG naznačen neznatno smanjenje amplitude a-vala i naglašeno smanjenje b-valova, tj lika pohranjene negativni negativne ERG. Kod nekih metala refleksne oftalmoskopski može se promatrati u središnjem dijelu mrežnice i periferije. Vidna oštrina može biti znatno smanjena. Lom može biti i emmetropic i ammetropicheskoy.
Tip Schubert-Bornstein je naslijedio u recesivni sex-linked tipa.
VIDEO: Pogled na centar "mikrokirurgija", mrežnice oka patologija
Kongenitalna stacionarni noćno slijepilo s promjenama fundus
Ogushi bolesti (Oguta, Oguchi) - Bilateralna nasljedna bolest koja se temelji na opsežnim degenerativnim promjenama šipke i čunjeva, bez uključivanja u patološkom procesu pigmentnog epitela mrežnice. Nasljeđivanje - autosomnoretsessivnoe. To utječe uglavnom japanskog podrijetla.
Uzrok bolesti se smatra da se zaustavi razvoj fotoreceptorske oka jedinice na razini normalne 7-8-mjesečnog fetusa.
Bolest se očituje u ranom djetinjstvu je gotovo potpuna sljepoća noću, smanjen oko adaptacije u sumrak i boja poremećaj vida. Oftalmoskopija napomenuti pepeo sivi fundus s metalik sjajem, tamnu nijansu žile na fundus periferiji, relativno tamne makularna područje žućkaste mrlje na očnu dne- pozitivan simptom Midzuo (obnavljanje normalnog fundusa boje nakon što je u mraku). Ove promjene se javljaju uglavnom u području ekvatora. Vidna oštrina ostaje normalan ili malo smanjio, periferni vid u granicama normale. Smanjenje tamni i maksimuma (zbog komponentu šipkasti) ERG Photopic ERG normalno. pigment svojstva ne mijenjaju.
Postoji nekoliko vrsta bolesti. Tipa I. bifazični tamna adaptacija krivulja promatrane tijekom duljeg boravka u mraku (2-3 do 24 sati), a tip II tamno adaptacija krivulja monofazna prirode.
Histološki otkrivaju veliku degeneraciju šipke i kukova.
Belotochechnoe fundusa karakteriziran izgled paramakulyarno i sredine periferne mrežnice multiple bijela žarišta u obliku točkastih podigne mjesta, koji se nalaze u dubljim slojevima mrežnice. Kada PAH otkriti više malih fenestrirane nedostatke na pigmentnog epitela u srednjem periferije mrežnice, ali za razliku od druz giperflyuorestsentsii ti nedostaci ne poštuju. Na dugo ostati u tamnim, tamnim praga prilagodbe može doseći normalnu razinu, iako u nekih bolesnika, čak iu tom slučaju, oni ostati povišen. Tamni ERG može biti normalan ili ispod normalnog negativni negativni. Tijekom dužeg tamno adaptacija ERG amplituda je ispravan. Karakteristično usporavanje regeneraciju vidnog pigmenta. To može biti povezana s alportov sindrom.
Histološko ispitivanje razotkrilo je fustsina nakupljanje granula u stanicama pigmentnog epitela i atrofija njegovih dijelova. Tip nasljeđivanja - autosomno recesivno.
Pjegavi mrežnica Iskrenost karakterizirana pojavom u dubokim slojevima mrežnice višestrukog žarišta nepravilnog oblika s jasnim granicama prljavo žute. Promjene su lokalizirani uglavnom na području ekvatora. Kada PAH događa u fokija giperflyuorestsentsiya. Photopic ERG nije promijenilo. Amplituda tamni ERG smanjena. Međutim, nakon dugog boravka u mraku, amplituda tamni ERG i krivulja tamne prilagodbe postiže standarde. Baš kao što je u fundus belotochechnom kinetiku vizualnih pigmenata smanjuje. Vidna oštrina i vizija boja nisu promijenili. Nasljeđivanje javlja u autosomno recesivno.
Progresivna chorioretinal distrofija
Tapetoretinalnoy retine abiotrophy (sin. Retinitis pigmentosa)
Morfološki otkrivenih uništavanje šipku i membranu s vakuolizacije vanjskim dijelovima, i nuklearne nestanak pleksiformni slojeva, migraciju pigment stanica u unutarnjim slojevima mrežnice, proliferacija glija stanica i vlakana koji ispunjavaju napustio prostor, fibroza i glioza žile retine i koroida, koroidalnu atrofija.
Više različitih oblika bolesti.
1. Genetski vrste:
- autosomno dominantni obrazac 9-43% svih slučajeva pigmentni degeneracije mrežnice. Karakteristično kasno pojavljivanje simptoma, sporog napredovanja;
- autosomno recesivno - najčešći oblik bolesti (20-35% svih slučajeva). Karakteristično rano početi;
- X-vezana tip - je najteži oblik retinitis pigmentosa, razvija u ljudima i dovodi do potpunog gubitka vida u četvrtom desetljeću. Žena, nositelj bolesti često imaju znakove blagog mrežnice lezije. Ovaj obrazac se nalazi u 8-45% slučajeva;
- sporadična vrsta je pronađena u 23-48% slučajeva.
2. Anatomski vrste:
- Tipičan - tipično za difuzne prirode promjena (hipo-pigmentacije i pigment migracija počinje vsredneekvato-ble zonu, postupno širi do centra). Promjene u makuli, mali, iako je u kasnijim fazama može uzrokovati degeneraciju „bika oka”;
- atipične - može nastaviti na nekoliko načina:
- difuzne promjene mogu se kombinirati s početkom patološki proces koji uključuje područje makularna;
- sektorski promjene - nakupine pigmenta u obliku polumjeseca na dnu u predjelu;
- jednostrani retinitis pigmentosa - Bilateralna asimetrična karakteristika lezije kasno u patološki proces koji uključuje drugom oku.
3. Funkcionalne poremećaji:
- Jednostavno progresivni tip - tip nasljeđivanja je obično dominantan. Karakterističan sporog napredovanja očuvanje vidne oštrine i do 60 godina;
- umjerene težine kod napredovanja u uznapredovalom stadiju - naznačen recesivno posjeda. Za 60 godina u razvoju vizije cijevi i smanjen vid;
- grubo napredovanje - karakteristika X-vezani načinu nasljeđivanja. Gubitak vidne funkcije postoji već 40 godina.
Očni simptomi. Smanjen tamno adaptacija. Tu je karakteristična uniforma prstenasta suženja granice periferna vidno polje do formiranja cijevnih. Nadalje, može biti kružni, skotomu, središnje paracentral scotomas ili sektorski (s središnja ili sektorskim oblicima bolesti) - postoji značajno smanjenje ili odsustvo ERG.
Za bolest karakterizirana taloženje pigmenta u obliku „tijela” kostiju i smanjuje količinu suženja mrežnice krvnih žila, poput voska blijedo optički disk. Na početku bolesti na periferiji mrežnice od ekvatora postoje područja hipo- i depigmenatciju, kao i pod-i intraretinalno bjelkaste sivkastu trikove i parcele sa crvenkastim boja. Zatim tu točku i granulirane inkluzija ( „sol i papar”), a zatim formira karakterističan oblik koštane stanice. Postupno, promjene se prenose s periferije prema centru. Proces obično ima dvosmjerni simetrični.
Modul} {direkt4
Promjene u makularne regiji pojaviti u 63-74% bolesnika. Najčešće nastaju: stushevannost i nestanak foveal refleksa, epiretinalna membrana atrofije i hypopigmentation (u kasnijim fazama), u zrnatost ili oskudno pigmentnog epitela, barem - distrofije tipa „bikovi očiju”, edem makule, cistična degeneracija i neke druge promjene.
Karakterizira kombinaciji sa kratkovidnosti, katarakt progresijom mrežnice promjena.
Kongenitalna Leberova kongenitalna slijepilo (syn. Tapetoretinalnoy djeca Leberova kongenitalna slijepilo)
Nasljedna bolest koja se manifestira bilateralne retinitis retinitom- trenutno smatrati disnukleotsidoz s uništavanjem šipke i kukova. Tip nasljeđivanja - autosomno recesivno, iako se u nekim slučajevima obilježen dominantnog nasljeđivanja.
Klinički znakovi i simptomi. Oftalmološki promjene u kombinaciji s mentalnom retardacijom, gubitak, različitih neuroloških poremećaja, uključujući napadajima sluha.
Histološki otkrivaju izolirana uništavanje pigmentnog epitela i odsutnosti fotoreceptorski sloja, umjesto kojeg se nalazi sloj kuboidnih stanica.
U tipičnom tijeku bolesti je vrlo niska vizija od rođenja. Djeca su plutajući pokrete očiju, pokreta grčevit oka, učenik inertan, bez pričvrsne stavke. U ranog djetinjstva tamo dan-sljepoće. Obično unutar prve godine života promjene u fundusu nisu dostupni, u starijoj dobi na periferiji dnu oka otkriti pigmentne naslage u obliku „koštanih stanica.” U uznapredovalim stadijima bolesti pojavljuju na retina raspršili pigmenta „prašinu” u normalnom optički disk ili pigmenta displazije makularna degeneracija ili mrežnice pigment fenomen pigmenta „kost tijela”, stanjivanje i bljedilo NHCONH plovila. Lom najviše dalekovidan sa psevdootekom optičkog diska, barem - kratkovidni. Karakterizira odsutnost ERG. Uočeno astigmatizam, naizmjenično konvergentni iskosa. Tu je duboko položaj u orbiti oko zbog retinalne atrofije, retrobulbami.
Laurence-Moon sindrom Bardet-Biedl (sin. Dientsefaloretinalnaya degeneracija)
Rijetka nasljedna bolest, koja se temelji na sekundarnom oštećenja mrežnice neuroepitelne abiotrophy i hipotalamo-hipofiza sustav. Tip nasljeđivanja - autosomno recesivno.
Klinički znakovi i simptomi. Bolest se očituje u prvoj godini života postupno pretilo gipogenitalizme, mentalna retardacija, polydactyly. Moguće koštane deformacije, syndactyly, ravna, gluhoća i gluho-turricephaly, patuljastog rasta ili gigantizma, zubni anomalije, bolest srca i bubrežna displazija hipoplazije, nefroskleroza, hidronefroza, pijelonefritisa, analni atrezija. Kod muškaraca, tu su impotencija i Azoospermia, ginekomastija, kriptorhizm- žene - vaginalni atrezija, Hipoplastični jajnici, oligo-ili amenoreja. Kao rezultat hormonalnih poremećaja razine metaboličkih procesa se smanjuje. Ponekad postoji neurološki poremećaj. Očekivano trajanje života ne prelazi 40 godina.
Morfološki utvrditi malformacije mozga - atrofiju vijugama agenezom corpus callosum, asimetrije hemisfera, hidrocefalus. Jezgre hipotalamusa izložak degeneracija sa smanjenjem broja ganglijskih stanica i rast vezivnog tkiva.
Očni simptomi. Karakterizira razvoj bilateralne pigmentna degeneracija mrežnice, koja se očituje u različitim kliničkim oblicima, kao i noćno sljepilo i smanjene viziju različitim stupnjevima. Označene degenerativne promjene u području makularne loma pogreške, strabizam (obično konvergira) microphthalmia, katarakta, atrofija očnog živca. Promjene u fundusa karakterizira visoka varijabilnost: mali pigmentirane žarišta okruglog ili nepravilnog oblika, razbacane dnu oka (tipičan oblik bolesti) - tipičan je pigmentna degeneracija uz taloženje pigmenta u obliku „kosti tijela” u periferiji optičke DNA- manje pigmentna degeneracija bez pigmenta- degeneracija mrežnice ponekad javlja u više žarišta bez bijelog pigmenta, razbacane donjem oka - degeneratio mrežnice punctata albescens.
Diferencijalna dijagnoza provesti sa sindromima Alstrema-Halgrena, Graefe, Usher, Prader-Willi, akrotsefalopolisin-daktil.
Horioderemiya
To se odnosi na nasljedne bolesti u kojima postoji simultano poraz mrežnice i choroid. X-vezani karakterističan naslijeđe. Bolest se javlja u polovini sinova žena - nositelja gena. Žene nositelji promjena gena u fundus blaga, oštrina vida se ne mijenja. Histološki pregled otkrio promijeniti pozicije povoljan pigmentnog epitela i fotoreceptora. Choroid na području nedostatka pigmentnog epitela također promijenila.
Očni simptomi. Karakterizirana pojavom u ranim poremećaja u djetinjstvu i tamne adaptacije vidnom polju promjena. Vidna oštrina je značajno smanjena na 30 godina. Na početku bolesti postoje višestruki skotomu i slijepa točka za proširenje. Postupno koncentrični sužavanje vidnom polju dovodi do stvaranja cijevi. Boja je vizija normalno ili postoji acyanopsia i tritanomaliya, ERG nije registrirana ili ima mikroERG.
Lagano sužavanje arterija, vena se ne mijenjaju. Četvrtina bolesnika dolazi s bljedilo od optičkog diska.
Atrofija girate (sin. Lobularni atrofija)
Odnosi se na bolesti koje imaju opću atrofije mrežnice i koroide. Razvoj bolesti uzrokovane nedostatkom enzima ornitinaminotransferazy. Tip nasljeđivanja - autosomno recesivno. Postoje dvije genetske forme: osjetljive i neosjetljive na vitamin B6. Bolest nastaje mutacija koje se nalaze na X kromosomu.
Klinički znakovi i simptomi. U krvi i drugim tjelesnim tekućinama povećanu razinu ornitin. Krvnoj plazmi smanjena razina glutamata, glutamina, kreatinina i Licinije. Nakon što je bliži degeneraciju skeletnim mišićima, promjene u EEG i EKG. Obilježje je prisutnost bolesnika s rijetkim ravnu kosu.
Očni simptomi. Promjene u fundusu pojaviti u prvom desetljeću života i postupno proširila iz periferije prema centru. Karakterizira pojavom žarišta atrofije s rubova scalloped. U ranoj dobi je noćno sljepilo i umanjena periferni vid. EWG nije registriran.
Pjegavi mrežnice abiotrophy
Alport Syndrome (sin. GR-oculo-bubrežni sindrom)
Nasljedna bolest, koja je određena dominantan gen, lokalizirano na X kromosomu. Suština sindroma progresivne bubrežne insuficijencije u kombinaciji s gluhoća.
Po obliku nasljedstva i kliničkih simptoma 6 vrsta sindrom: autosomno dominantno s klasičnim juvenilni tip gluhotoy- X-vezani recesivni gluhotoy- s X-vezani recesivni odrasle gluhotoy- sindrom X-vezani recesivni gluhoće bez odraslih sindrom i druga defektov- autosomno -dominantny sindrom gluhoće i autosomno trombotsitopeniey- retsissivny juvenilni sindrom s gluhoća,
Klinički znakovi i simptomi. Bolest se može manifestirati u ranom djetinjstvu gubitka Senzorineuralna sluha, razvojni poremećaji bubrega i uretera s hematurija, proteinurija, bakteriurija. Zatajenje bubrega javlja češće u muškaraca - dječaka obično umiru tijekom adolescencije.
simptomi oka javljaju u 1/6 bolesnika i očigledne abnormalnosti tijelo prednje ili stražnje lentikonus- prednja i stražnja supkapsularnu katarakta- simptom Krukenberga- degenerativnim promjenama mrežnice u obliku brojnih bjelkaste tochek- sferofakiya, mikrofakiya.
Video: Otvaranje dječjeg ured za zaštitu
Biett distrofije (sin. Mrežnice abiotrophy ruba rožnice distrofija)
Kompleks simptomi očne dystrophic. Nasljedna bolest, rijetka, sporo progresivna, a poraz na oba oka. Etiopatogeneza je nepoznat. Pustite, muškarci i žene svih dobi.
Očni simptomi. Bolest se razvija u 3-4th desetljeća života, manifestira u obliku rožnice lezija granične degeneracije. Stromu rožnice kod limba pojaviti sjajna, žućkasto igala i zapečen mjesta. Distrofična zatim razvija u proces choroid i mrežnice, naznačen sklerotičan lezije su formirane u obliku okruglih točaka ili „tijela” kosti koje daju oka donji smeđe. retinalne vaskularne sužavanje je manje izražena nego kod drugih oblika degeneracije. NHCONH dekolorirovan. Funkcionalni poremećaji su definirani kao nedostataka u vidnom polju, noćno sljepilo, progresivni suženjem vizualnog polja prije formiranja cijevi, te značajnog smanjenja vidne oštrine.
Diferencijalna dijagnoza provesti s retinitis pigmentosa.
Druze staklen korioidea ploča
Rijetka nasljedna bolest učestalost koja je 0.35 na 1000 pacijenata s očne patologije. Karakteriziraju smetnja normalnog metabolizma u intermedijera mrežnice pigment tvari i samih epitelnih stanica. Tip nasljeđivanja - autosomno dominantna s promjenjivom pojavnosti i ekspresivnosti.
patogeneza. Kao rezultat metaboličkih poremećaja u mrežnici se nakupila u citoplazmi stanica pigmentnog epitela govore hijalina netopiva Proteinski materijal. Tu proteinosis pojedine stanice ili skupine stanica epitela. Kvržice govore hijalina tvar spajanje, njihov uzgoj obložene pigment epitelnih stanica susjedna zdrava područja. Produkt je pigment epitelne stanice - glamelinovye tijelo - se Druz staklastu ploču. Oni leže na staklene ploče (Bruchove membrane), koji je oštro razgraničena od njih. U velikim druza stanicama imaju vretenastom ili ravan oblik s povremenim jezgre i malom količinom pigmenta zrna. On sadrži netopivi kalcijeve soli. Kasnije se razvija okoštavanja prijatelje.
Očni simptomi. Ophthalmoscopically druze pojavljuju kao žućkasta ili bjelkasto-žućkasti žarišta sa zaobljenim oštrih rubova, veličine obično nije veći od dvostrukog promjera vene na NHCONH. Druz često nalazi u temporalnom dijelu optičkog diska i oko makularne području. Oni se nalaze na unutarnjoj površini Bruchove membrane u obliku malih žarišta. Ponekad narasti. U tim slučajevima, vidljivu oko ruba pigmenta koji se sastoji od proliferirajućih RPE stanice.
Video: 2015/11/08 - Program "Trideset i šest godina i šest" (36.6)
Koloidna degeneracija mrežnice iskrenosti
Dystrophic bolest mrežnice koja pogađa žene u dobi od 30-40 godina. Temeljne bolesti su primarni degeneraciju Bruchove membrane i degeneraciju pigmenta epitelne stanice.
Očni simptomi. Bolest počinje sa smanjenjem tamne prilagodbe. Nakon toga, kako bolest napreduje u drugom stupnju se smanjuje periferni i centralni vid. Ophthalmoscopically u regiji pokazuju paramakulyarnoy vrlo velika (do 1 / W DD) druze plosnate nepravilne izduženog oblika. Pomicanjem središte udaranje zone makularna druze napredovanja procesa za nekoliko godina.
Sjogrenov sindrom-Larsson
Nasljedne bolesti degenerativnog prirode, karakteriziran trijade simptoma: kongenitalnog ihtioze, mentalne retardacije i udova spastički tetraparesis. Patogeneza nije jasno. Neki istraživači su povezali razvoj bolesti s povećanjem nekih bolesnika mokraći kinurenin i histidin. Tip nasljeđivanja - autosomno recesivno.
tijekom progresivne bolesti, stabilne ponekad. Prognoza sve nepovoljne.
Očni simptomi. Patološke promjene može doći do oka Ectropion, stanjivanje rožnice, formiranje erozija i ulkusa rožnice sa svojim mogućim perforacije. Mali udio bolesnika sa sindromom očituje vrsta pigmenta degeneracije kod male bijele žarišta u središnjem području.
 Klasifikacija ahondrogeneza fetus. dijagnoza ahondrogeneza
Klasifikacija ahondrogeneza fetus. dijagnoza ahondrogeneza Osteogenesis imperfecta fetus. Uzroci i dijagnoza osteogenesis imperfecta
Osteogenesis imperfecta fetus. Uzroci i dijagnoza osteogenesis imperfecta Fibrohondrogenez i atelosteogenez. Ahondrogenez ili anosteogenez
Fibrohondrogenez i atelosteogenez. Ahondrogenez ili anosteogenez Osteogenesis imperfecta. Dijagnoza i prognoza osteogenesis imperfecta u fetus
Osteogenesis imperfecta. Dijagnoza i prognoza osteogenesis imperfecta u fetus Dominantnih i recesivnih alela kromosoma. Autosomno dominantnih nasljedstvo
Dominantnih i recesivnih alela kromosoma. Autosomno dominantnih nasljedstvo Autosomno recesivno nasljeđivanje. X-vezani nasljeđe
Autosomno recesivno nasljeđivanje. X-vezani nasljeđe Galaktoziltseramidoz Krabbe bolest. Sulfatidoz metakromatskom leykodistrosriya Scholz
Galaktoziltseramidoz Krabbe bolest. Sulfatidoz metakromatskom leykodistrosriya Scholz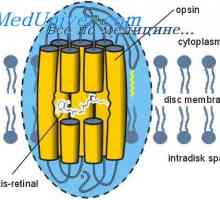 Svjetlo i tama adaptacija. Mehanizmi svijetlih i tamnih adaptaciju
Svjetlo i tama adaptacija. Mehanizmi svijetlih i tamnih adaptaciju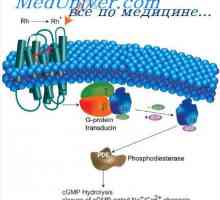 Sljepoća na pojedine boje. Funkcija neurona retine
Sljepoća na pojedine boje. Funkcija neurona retine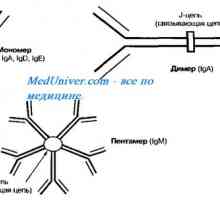 Autosomna recesivna sindrom hiperprodukcija imunoglobulina m (IgM). genske mutacije pomoć
Autosomna recesivna sindrom hiperprodukcija imunoglobulina m (IgM). genske mutacije pomoć Neke funkcionalne metode istraživanja
Neke funkcionalne metode istraživanja Dan-sljepoća (noćno sljepilo, noćna sljepoća) -rasstroystvo sumrak vizija. Etiologija kongenitalne…
Dan-sljepoća (noćno sljepilo, noćna sljepoća) -rasstroystvo sumrak vizija. Etiologija kongenitalne…- Katarakta- tvari ili zamućenje kapsule leće. Etiologija: metabolički poremećaji, toksični i…
- Zdravlje Enciklopedija, bolest, lijek, liječnik, apoteka, infekcija, sažetak, seks, ginekologije,…
- Zdravlje Enciklopedija, bolest, lijek, liječnik, apoteka, infekcija, sažetak, seks, ginekologije,…
- Zdravlje Enciklopedija, bolest, lijek, liječnik, apoteka, infekcija, sažetak, seks, ginekologije,…
 Albinizam kod ljudi, oka
Albinizam kod ljudi, oka Goniodisgenez
Goniodisgenez Sindromi, uključujući i patologije staklovine
Sindromi, uključujući i patologije staklovine Kongenitalne makularna promjene
Kongenitalne makularna promjene Monogenskih sindromi mvpr
Monogenskih sindromi mvpr
 Galaktoziltseramidoz Krabbe bolest. Sulfatidoz metakromatskom leykodistrosriya Scholz
Galaktoziltseramidoz Krabbe bolest. Sulfatidoz metakromatskom leykodistrosriya Scholz Fibrohondrogenez i atelosteogenez. Ahondrogenez ili anosteogenez
Fibrohondrogenez i atelosteogenez. Ahondrogenez ili anosteogenez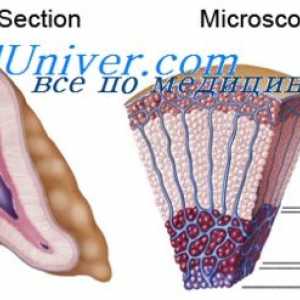 Osteogenesis imperfecta. Dijagnoza i prognoza osteogenesis imperfecta u fetus
Osteogenesis imperfecta. Dijagnoza i prognoza osteogenesis imperfecta u fetus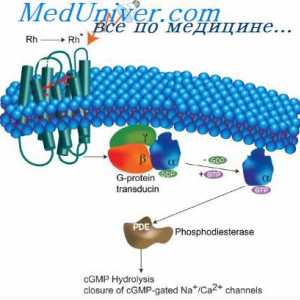 Sljepoća na pojedine boje. Funkcija neurona retine
Sljepoća na pojedine boje. Funkcija neurona retine Klasifikacija ahondrogeneza fetus. dijagnoza ahondrogeneza
Klasifikacija ahondrogeneza fetus. dijagnoza ahondrogeneza Goniodisgenez
Goniodisgenez